Sickle cell anemia is an inherited condition of the globin chains that leads to hemolysis and chronic organ damage.
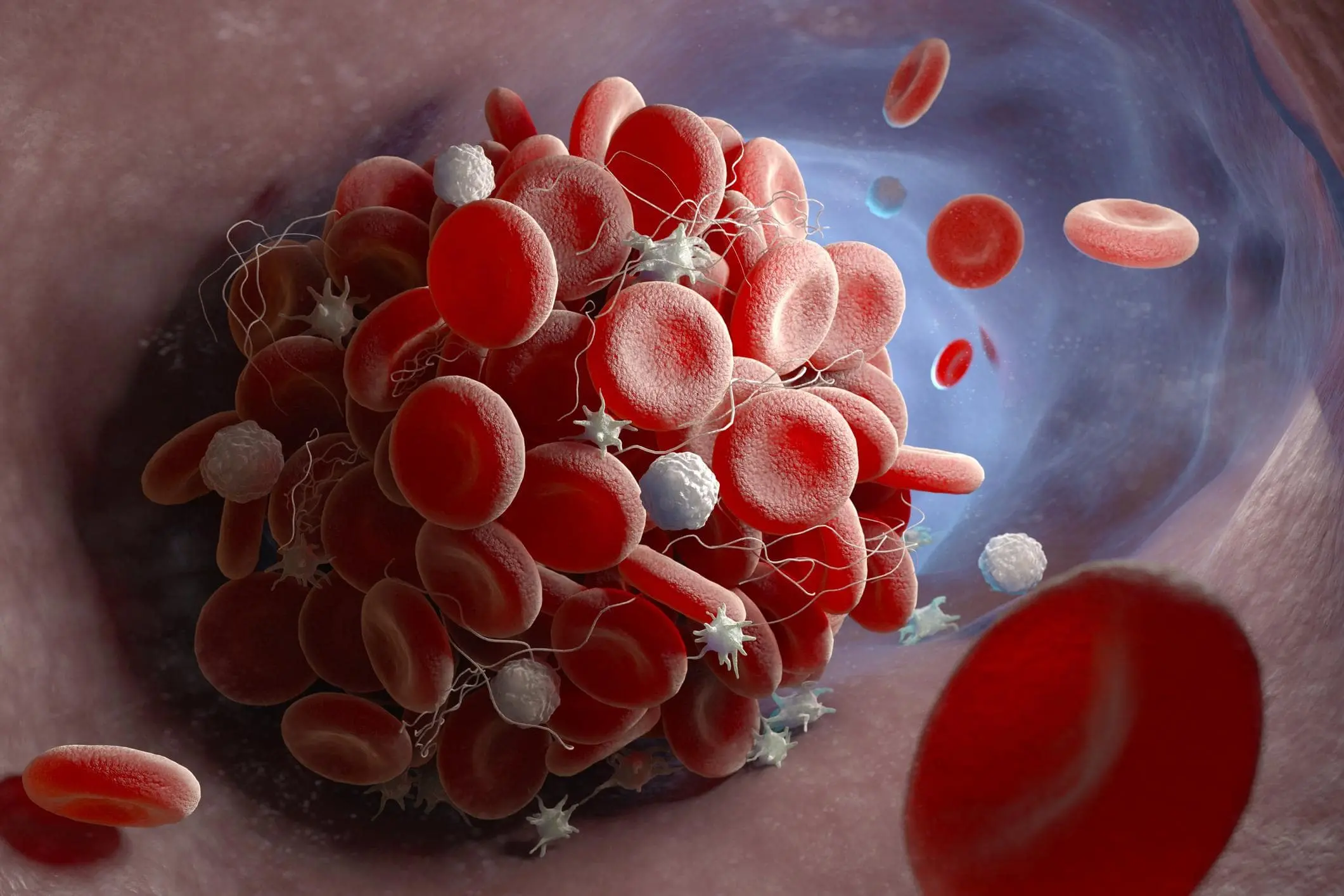
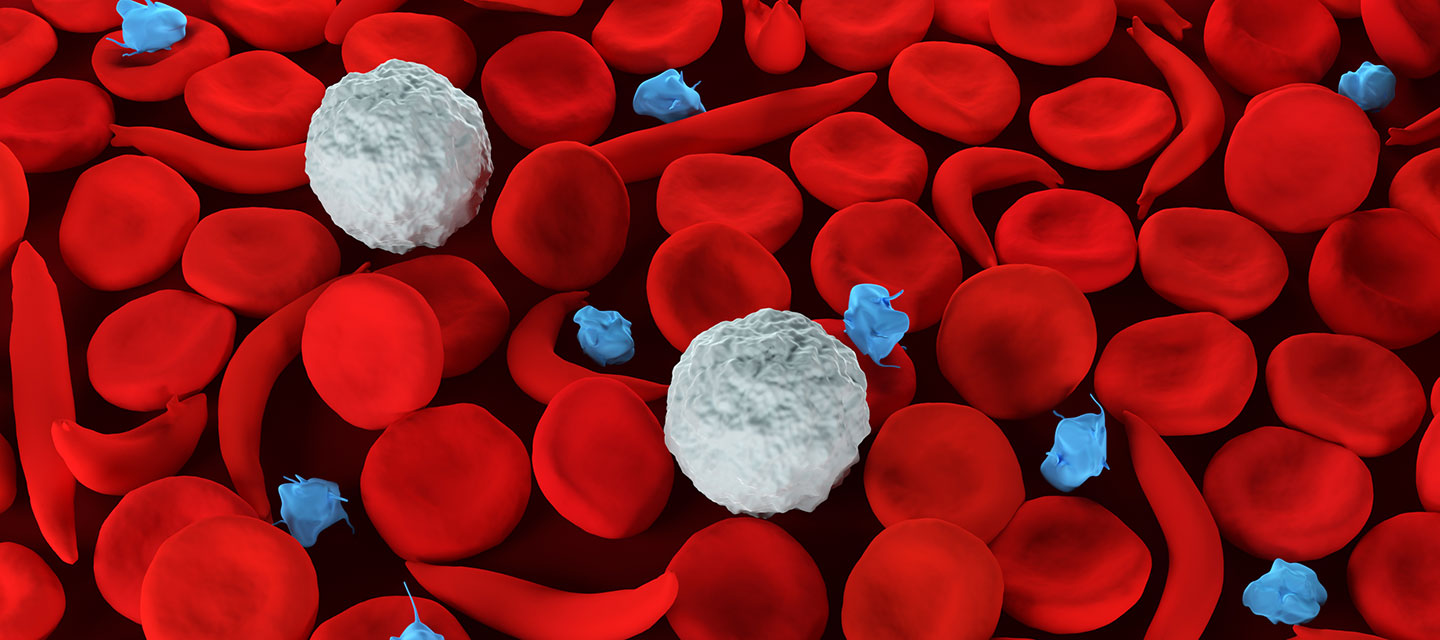
It is a chronic blood condition characterized by abnormally rigid and sickle-shaped red blood cells.
Hemolysis and Vaso-occlusive crises are the two primary characteristics of sickle cell anemia (VOC).
The gene defect is caused by a known mutation of a single nucleotide (A to T) of the p-globin gene, resulting in glutamate being replaced by valine at position 6.
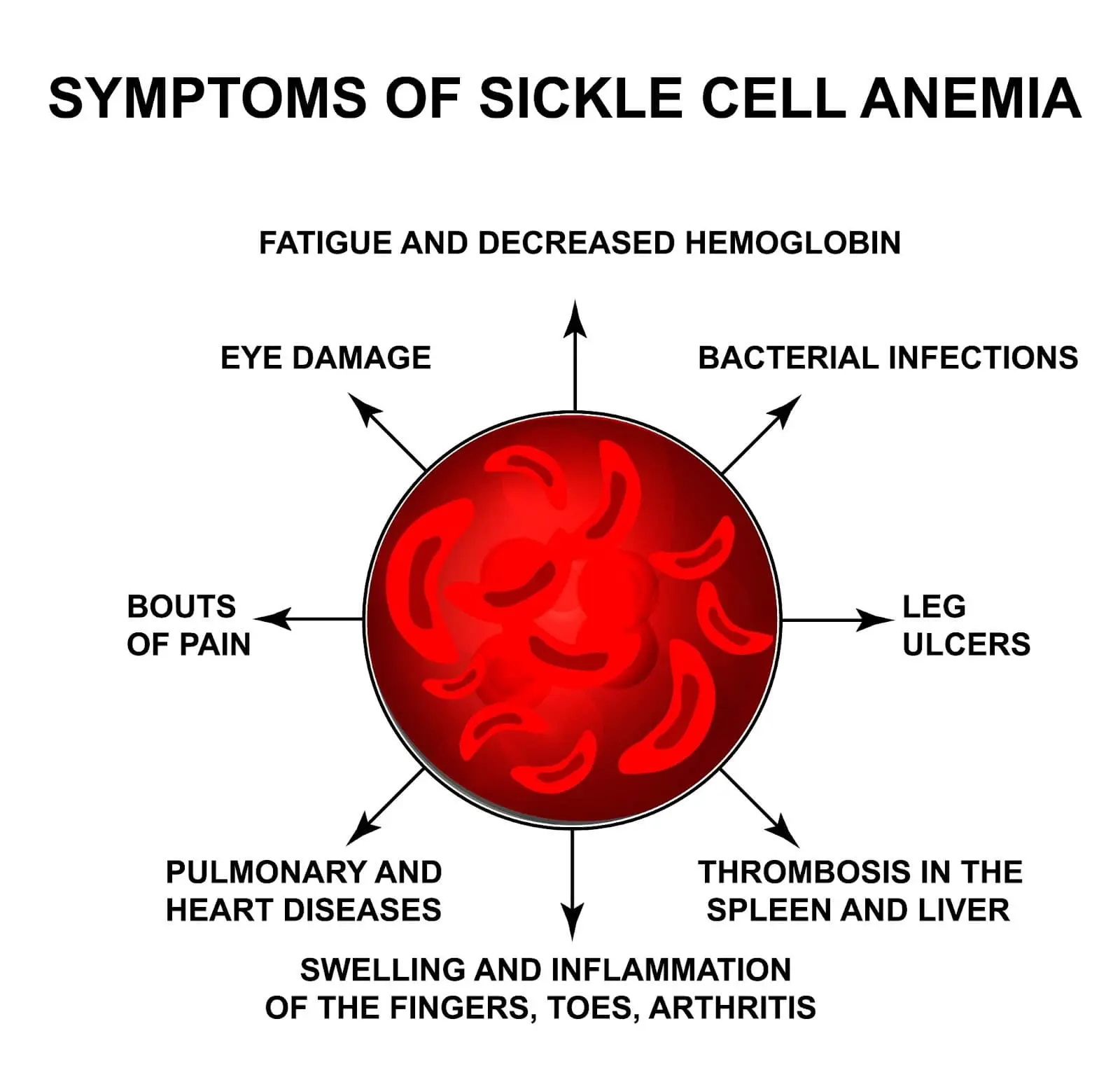
Sickle cell anemia manifests as severe hemolytic anemia that is periodically interrupted by crises.
The most common type of crisis is a Vaso-occlusive crisis.
Their etiology is usually attributed to low oxygen tension as in high altitude, water loss, and infection.
Vaso-occlusion causes intense pain, particularly in the bones (hips, shoulders, and vertebrae).
Infarcts of the small bones cause painful dactylitis (hand-foot syndrome).
The complete blood count in HbSS reveals hemoglobin levels in the range of 6-8 g/dL, with reticulocytosis (due to compensatory bone marrow hyperplasia).
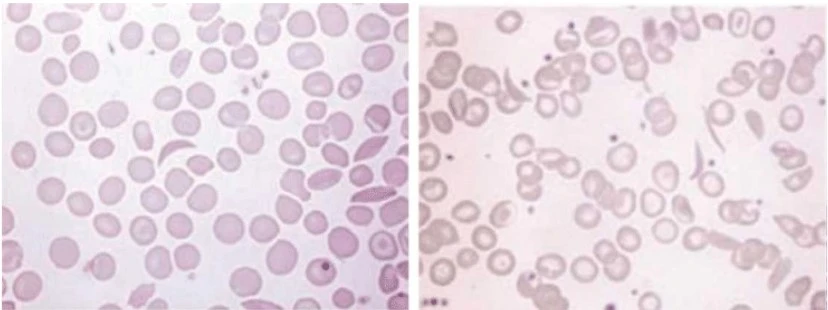
A blood film may show sickle-shaped cells and hyposplenism characteristics (target cells and Howell-Jolly bodies).
Confirmation of the diagnosis is accomplished using high-performance liquid chromatography (HPLC).
When compared to normal adults, sickle cell disease has a lower overall life expectancy (perhaps by 20 to 30 years), but advances in therapy are extending survival.
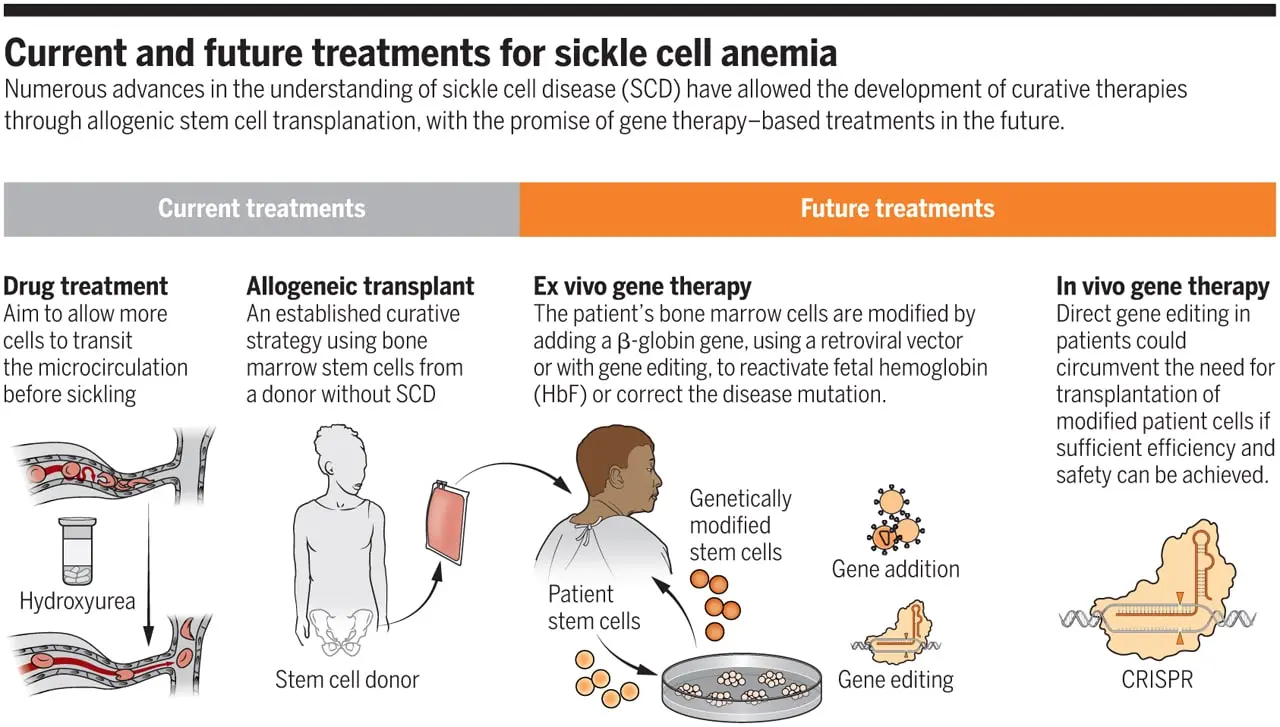
Voxelotor and crizanlizumab are two newer medications that were approved in 2019, also the increased use of hematopoietic stem cell transplants, and research into novel methods like gene therapy all point to an improvement in quality of life and survival rates.
Treatment aims is to prevent RBC sickling, dehydration, hypoxia, and acidosis, all of which can induce sickling.
The typical approach to managing a crisis is supportive unless a blood transfusion is necessary.
For the management of severe pain attacks, it is frequently necessary to administer subcutaneous morphine or another strong opiate.
Children born with sickle cell disease will be given folic acid (1 mg dose) every day for the rest of their lives.
Patients must also take penicillin every day from birth until age five due to their susceptibility to pneumococcal infection.
Sickle cell disease has a great chance of being cured with hematopoietic stem cell transplantation (bone marrow transplantation).